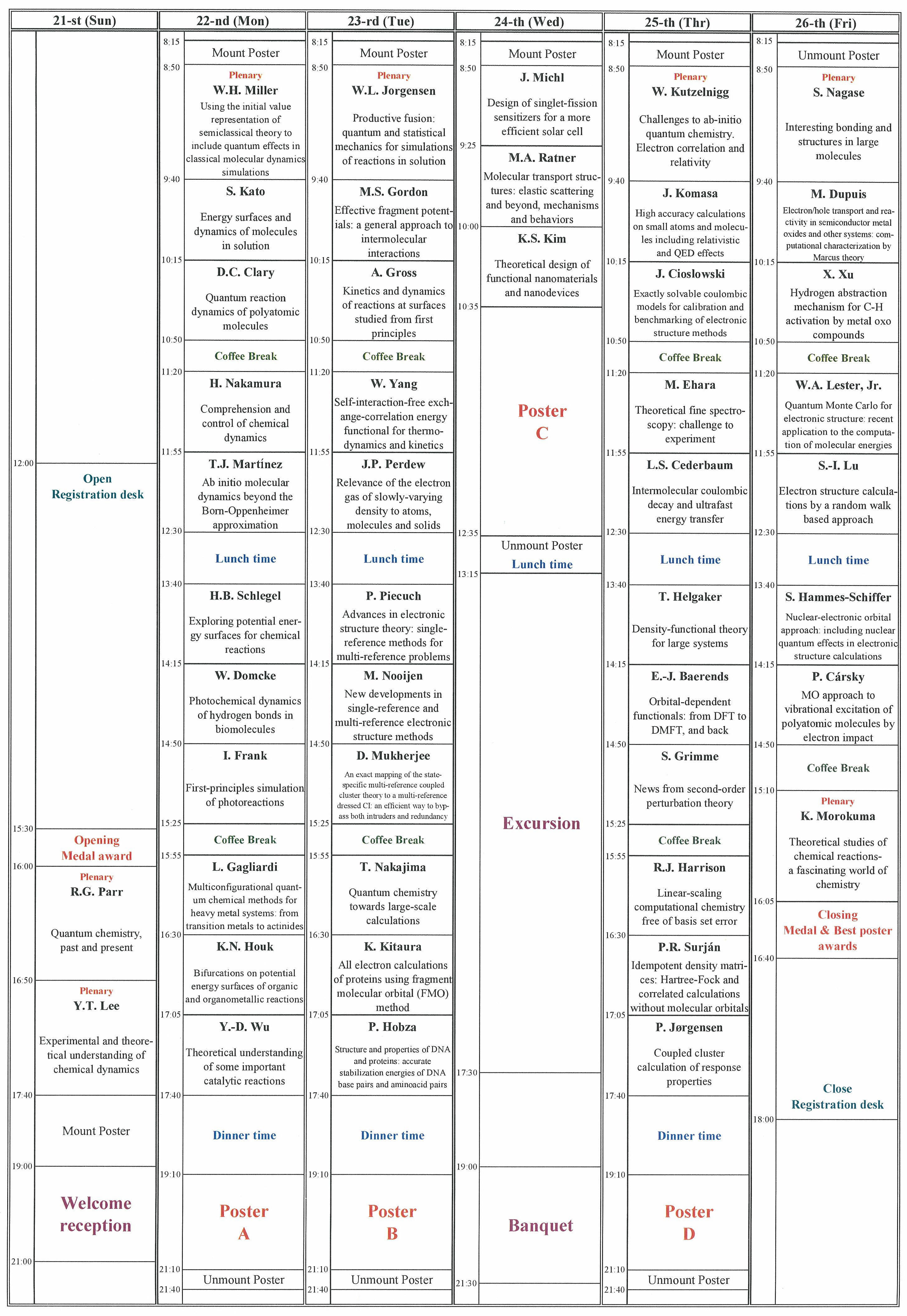
|
| Evert-Jan Baerends |
Vrije Universiteit, The Netherlands |
Orbital-dependent functionals: from DFT to DMFT, and back |
|
| Petr Cársky |
Academy of Sciences of the Czech Republic, Czech Republic |
MO approach to vibrational excitation of polyatomic molecules by electron
impact |
|
| Lorentz S. Cederbaum |
Universitaet Heidelberg, Germany |
Intermolecular coulombic decay and ultrafast energy transfer |
|
| Jerzy Cioslowski |
University of Szczecin, Poland |
Exactly solvable coulombic models for calibration and benchmarking of electronic
structure methods |
|
| David C. Clary |
Oxford University, UK |
Quantum reaction dynamics of polyatomic molecules |
|
| Wolfgang Domcke |
Technische Universitaet Muenchen, Germany |
Photochemical dynamics of hydrogen bonds in biomolecules |
|
| Michel Dupuis |
Pacific Northwest National Laboratory, USA |
Electron/hole transport and reactivity in semiconductor metal oxides and
other systems: computational characterization by Marcus theory |
|
| Masahiro Ehara |
Kyoto University, Japan |
Theoretical fine spectroscopy: challenge to experiment |
|
| Irmgard Frank |
Universitaet Muenchen, Germany |
First-principles simulation of photoreactions |
|
| Laura Gagliardi |
University of Geneva, Switzerland |
Multiconfigurational quantum chemical methods for heavy metal systems:
from transition metals to actinides |
|
| Mark S. Gordon |
Iowa State University, USA |
Effective fragment potentials: a general approach to intermolecular interactions |
|
| Stefan Grimme |
Universitaet Muenster, Germany |
News from second-order perturbation theory |
|
| Axel Gross |
Technische Universitaet Muenchen, Germany |
Kinetics and dynamics of reactions at surfaces studied from first principles |
|
| Sharon Hammes-Schiffer |
Pennsylvania State University, USA |
Nuclear-electronic orbital approach: including nuclear quantum effects
in electronic structure calculations |
|
| Robert J. Harrison |
Oak Ridge National Laboratory, USA |
Linear-scaling computational chemistry free of basis set error |
|
| Trygve Helgaker |
University of Oslo, Norway |
Density-functional theory for large systems |
|
| Pavel Hobza |
Academy of Sciences of the Czech Republic, Czech Republic |
Structure and properties of DNA and proteins: accurate stabilization energies
of DNA base pairs and aminoacid pairs |
|
| Kendall N. Houk |
University of California, USA |
Bifurcations on potential energy surfaces of organic and organometallic
reactions |
|
| Poul Jørgensen |
Arhus University, Denmark |
Coupled cluster calculation of response properties |
|
| Shigeki Kato |
Kyoto University, Japan |
Energy surfaces and dynamics of molecules in solution |
|
| Kwang S. Kim |
Pohang University of Science and Technology, Korea |
Theoretical design of functional nanomaterials and nanodevices |
|
| Kazuo Kitaura |
National Institute of Advanced Industrial Science and Technology, Japan |
All electron calculations of proteins using fragment molecular orbital (FMO) method |
|
| Jacek Komasa |
A.Mickiewicz University, Poland |
High accuracy calculations on small atoms and molecules including relativistic
and QED effects |
|
| William A. Lester, Jr |
University of California, Berkeley, USA |
Quantum Monte Carlo for electronic structure: recent application to the
computation of molecular energies |
|
| Shih-I. Lu |
Fooyin University, Taiwan |
Electron structure calculations by a random walk based approach |
|
| Todd J. Martínez |
University of Illinois, USA |
Ab initio molecular dynamics beyond the Born-Oppenheimer approximation |
|
| Josef Michl |
University of Colorado, USA |
Design of singlet-fission sensitizers for a more efficient solar cell |
|
| Debashis Mukherjee |
Indian Association for the Cultivation of Science, India |
An exact mapping of the state-specific multi-reference coupled cluster
theory to a multi-reference dressed CI : an efficient way to bypass both
intruders and redundancy |
|
| Takahito Nakajima |
University of Tokyo, Japan |
Quantum chemistry towards large-scale calculations |
|
| Hiroki Nakamura |
Institute for Molecular Science, Japan |
Comprehension and control of chemical dynamics |
|
| Marcel Nooijen |
University of Waterloo, Canada |
New developments in single-reference and multi-reference electronic structure
methods |
|
| John P. Perdew |
Tulane University, USA |
Relevance of the electron gas of slowly-varying density to atoms, molecules
and solids |
|
| Piotr Piecuch |
Michigan State University, USA |
Advances in electronic structure theory: single-reference methods for multi-reference
problems |
|
| Mark A. Ratner |
Northwestern University, USA |
Electronic motion in molecular circuits: a problem in theoretical chemistry |
|
| H. Bernhard Schlegel |
Wayne State University, USA |
Exploring potential energy surfaces for chemical reactions |
|
| Péter R. Surján |
Eoetvoes University, Hungary |
Idempotent density matrices: Hartree-Fock and correlated calculations without
molecular orbitals |
|
| Yun-Dong Wu |
Hong Kong University, China |
Theoretical understanding of some important catalytic reactions |
|
| Xin Xu |
Xiamen University, China |
Hydrogen abstraction mechanism for C-H activation by metal oxo compounds |
|
| Weitao Yang |
Duke University, USA |
Self-interaction-free exchange-correlation energy functional for thermodynamics
and kinetics |
|